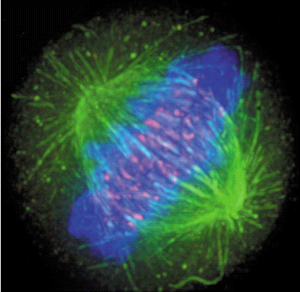
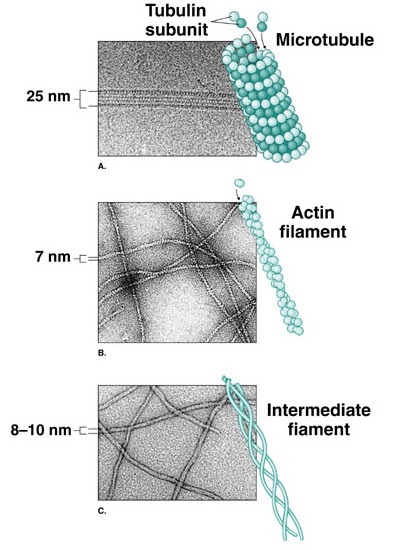
Nós aprendemos na escola que o responsável pela transmissão da informação genética é a molécula de DNA. Aliás, no CAp (Colégio da Aplicação da UFRJ), a minha “aula inaugural”, chamada de regência, foi sobre mitose e englobou também alguns conceitos envolvendo o DNA: cromossomos, cromátides, compactação do DNA, etc. A aula foi sobre como a célula se divide em duas, e como a sua célula-filha continua tendo o mesmo material genético e em igual quantidade que a célula-mãe. Mas isto será tema de outro post.
Voltando à transmissão da informação genética: é provável que algum aluno mais curioso tenha se perguntado: “mas como eles descobriram que era o DNA?”. Muito simples: fazendo experimentos.
Quem deu o pontapé inicial foi Frederick Griffith, nos anos de 1928 (Detalhe: O DNA já havia sido descoberto nessa época, só a sua estrutura molecular que foi elucidada por Watson e Crick. Pelo amooor de Deus, não foram eles que descobriram o DNA!!!). Ele trabalhava com dois tipos de uma bactéria pneumococus: uma encapsulada que causava infecção em ratos (portanto, era patogênica) e outra não encapsulada, que não causava infecção em ratos. (PS.: Algumas bactérias possuem uma cápsula em torno da sua parede celular). Ele identificava as colônias das bactérias a partir do aspecto das colônias: às não patogênicas chamou de R (do inglês rough, que significa rugoso) e às patogênicas, de S (do inglês smooth, que significa liso). Para facilitar a leitura do texto, vou criar uma legenda: Bactérias R são ñPat. (não patogênicas) e bactérias S são Pat. (patogênicas).

Esse é o F. Griffith
A partir disso, ele fez alguns experimentos. Injetou as bactérias S (Pat.) vivas em camundongos. Como era esperado, os ratos morreram. Em seguida fez o mesmo utilizando as bactérias R (ñPat.) e os ratos sobreviveram. Um terceiro experimento foi injetar as bactérias R (ñPat.) mortas pelo calor. Será que os ratos sobreviveram? Sim, sobreviveram, afinal as bactérias patogênicas foram mortas pelo calor. Então Griffith pegou os “restos” das baterias S (Pat.) mortas pelo calor e misturou com bactérias R (ñPat.) vivas e injetou esta mistura nos ratos. Aposto um doce como os ratos sobreviveram, alguém diria... mas não, os ratos NÃO sobreviveram. Para ter certeza dos resultados, Griffith repetiu várias vezes este mesmo experimento para ter certeza que tudo estava certo. Então ele verificou amostras de sangue dos ratos mortos no último experimento e encontrou as bactérias S (PATOGÊNICAS) vivas. Como foi possível, se antes ele havia injetado as bactérias S mortas pelo calor e os ratos sobreviveram? Havia alguma substância que transformou as bactérias do tipo R no tipo S.
E os responsáveis por desvendar este mistério foram 3 pesquisadores: Oswald Avery, Colin MacLeod e Maclyn MacCarty. O ano: 1944. Eles usaram as mesmas bactérias, só que em vez de injetar em ratos, eles faziam culturas delas em placas de Petri, além de trabalhar com extratos celulares destas bactérias (o extrato celular é o líquido que se obtém após romper as células, contendo todas as substâncias presentes). Eles meio que “repetiram” os experimentos de Griffith. Primeiramente, fizeram uma cultura das bactérias R (ñPat.). Em seguida, rompeu as bactérias S (Pat.), ou seja, fez um extrato celular, e adicionou em uma cultura para células. Como era de se esperar, não cresceu nada. Agora eles pegaram as bactérias R (ñPat.), misturaram com extrato celular das bactérias S (Pat.) e preparou a cultura. E as células que cresceram foram...as do tipo S (PATOGÊNICAS)! Viu como foi repeteco do experimento do Griffith? Agora vem a novidade: eles queriam descobrir qual molécula era responsável pela transformação da bactéria R em S. Então eles prepararam o seguinte experimento: misturaram às bactérias R (ñPat.) vivas o extrato da S adicionado de: 1)enzima protease (que degrada proteínas); 2) DNAse (que degrada o DNA) e 3) RNAse (que degrada o RNA). A idéia era a seguinte: se a proteína fosse responsável pela transformação da bactéria, se adicionarmos algo que destrua/acabe/inative com a proteína a bactéria não vai se transformar. Este raciocínio serve também para o DNA e RNA. E então, qual cultura não cresceu bactéria, a de nº 1, nº2 ou nº 3? Respostas no “meus comentários” deste blog, por favor... e com justificativa!
 Este é o Oswald Avery
Este é o Oswald Avery 







 E depois de pescados os cortes, as grades vão para o...porta-grades. Uma caixinha com vários buraquinhos numerados para guardar os cortes até a hora de usarmos...
E depois de pescados os cortes, as grades vão para o...porta-grades. Uma caixinha com vários buraquinhos numerados para guardar os cortes até a hora de usarmos...







 Figura 1: Hemócitos em microscopia de luz e C de fase interferencial. Em A, temos a hemoblasto-like. Em B, hialinócito. Em C, granulócito. Em G, um agregado de cél, que graças a Deus ñ tenho visto muito nestas placas que ando fazendo, pois antes tinha muuuito deste tipo. quando tem, são de poucas células. em H, granulócito degranulado.
Figura 1: Hemócitos em microscopia de luz e C de fase interferencial. Em A, temos a hemoblasto-like. Em B, hialinócito. Em C, granulócito. Em G, um agregado de cél, que graças a Deus ñ tenho visto muito nestas placas que ando fazendo, pois antes tinha muuuito deste tipo. quando tem, são de poucas células. em H, granulócito degranulado. Tabela 1: resumo da microscopia de luz
Tabela 1: resumo da microscopia de luz
 Tabela 2: Diferentes subpopulações de hemócitos classificados por citometria de fluxo
Tabela 2: Diferentes subpopulações de hemócitos classificados por citometria de fluxo





{kind=link}